Exp. No.7 Extraction and separation of chromosomal DNA from Bacteria
Extraction and separation of chromosomal DNA from Bacteria
Introduction
The
isolation and purification of DNA from cells is one of the most common
procedures in contemporary molecular biology and embodies a transition from
cell biology to the molecular biology (from in vivo to in vitro). The isolation
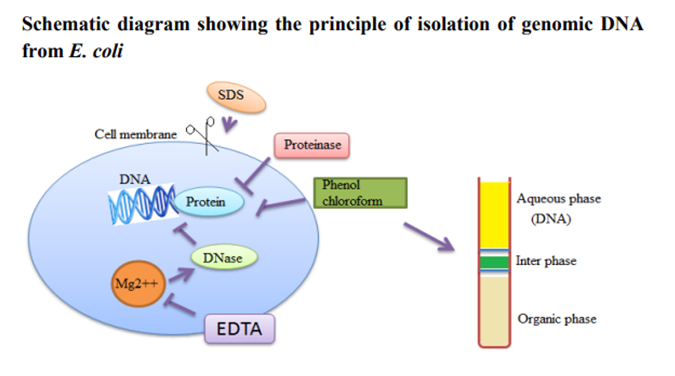
of DNA from bacteria is a relatively simple process. The organism to be used
should be grown in a favorable medium at an optimal temperature and should be
harvested in late log to early stationary phase for maximum yield. The genomic
DNA isolation needs to separate total DNA from RNA, protein, lipid, etc.
Initially the cell membranes must be disrupted in order to release the DNA in
the extraction buffer. SDS (sodium dodecyl sulphate) is used to disrupt the
cell membrane. Once cell is disrupted, the endogenous nucleases tend to cause
extensive hydrolysis. Nucleases apparently present on human fingertips are
notorious for causing spurious degradation of nucleic acids during
purification. DNA can be protected from endogenous nucleases by chelating Mg2++
ions. Mg2++ ion is considered as a necessary cofactor for action of most of the
nucleases. Nucleoprotein interactions are disrupted with SDS, phenol or
proteinase K. Proteinase enzyme is used to degrade the proteins in the
disrupted cell soup. Phenol and chloroform are used to denature and separate
proteins from DNA. Chloroform is also a protein denaturant, which stabilizes
the rather unstable boundary between an aqueous phase and pure phenol layer.
The denatured proteins form a layer at the interface between the aqueous and
the organic phases which are removed by centrifugation. DNA released from
disrupted cells is precipitated by cold absolute ethanol or isopropanol.
Aim
To isolate the genomic DNA from E. coli cells
Materials and Methods
LB Broth, E. coli
cells, Reagents, TE buffer (pH 8.0), 10% SDS, Proteinase K, Phenol-chloroform
mixture, 5M Sodium Acetate (pH 5.2), Isopropanol, 70% ethanol, Autoclaved
Distilled Water, Eppendorf tubes 2 ml, Micropipette, Microtips, Microfuge
Preparation of
Reagents:
1.
TE Buffer (pH 8.0): 10 mm TrisHCl (pH 8.0), 1 mm EDTA (pH 8.0)
·
For a 1 M solution, dissolve 12.1 g of
Tris base in 80 mL of nuclease-free water.
·
Adjust the pH to 8 values by slowly
adding approximately 6-7 mL concentrated HCl. Adding concentrated HCl to the
Tris buffer will increase the temperature of the solution, which affects the
pH. Allow the solution to cool to room temperature before making final
adjustments to the pH (using more HCl if necessary).
·
Adjust the volume of the solution to 100
mL with water
·
To obtain a 10 mM Tris-HCl pH 8
solution, dilute 1 M Tris-HCl 1:100 with nuclease-free water. For example, add
1 mL of 1 M Tris-HCl to 99 mL of nuclease-free water.
|
Reagent |
Volume |
Final concentration |
|
1 mL |
10 mM |
|
|
0.2 mL |
1 mM |
|
|
Distilled H2O |
98.8 mL |
2. 10% SDS: Dissolve 10 g of SDS in 100
ml autoclaved distilled water.
3. Proteinase K: Dissolve 10 mg of
Proteinase K in 1 ml autoclaved distilled water.
4. Phenol – Chloroform Mixture: The pH
is very important. For RNA purification, the pH is kept around pH 4, which
retains RNA in the aqueous phase preferentially. For DNA purification, the pH
is usually 7 to 8, at which point all nucleic acids are found in the aqueous
phase. Mix equal volume of phenol with chloroform. Keep the mixture on ice and
add 20 ml TE buffer, extract by shaking for 15 minutes. Remove the dust on the
surface layer using a pipette. Repeat 4-5 times. Add 30-40 ml of TE buffer and
store it on ice.
5. 5M Sodium Acetate: Dissolve 41 g of
sodium acetate in 100 ml distilled water and adjust pH with dilute acetic acid
(pH 5.2).
6. Isopropanol
7. 70% Ethanol
Procedure
·
2 ml overnight culture was taken and the
cells were harvested by centrifugation for 10 minutes
·
875 µl of TE buffer was added to the
cell pellet and the cells were resuspended in the buffer by gentle mixing.
·
100 µl of 10% SDS and 5 µl of Proteinase
K were added to the cells.
·
The above mixture was mixed well and
incubated at 37º C for an hour in an incubator.
·
1 ml of phenol-chloroform mixture was
added to the contents, mixed well by inverting and incubated at room
temperature for 5 minutes.
·
The contents were centrifuged at 10,000
rpm for 10 minutes at 4º C.
·
The highly viscous jelly like supernatant
is collected using cut tips and is transferred to a fresh tube.
·
The process was repeated once again with
phenol-chloroform mixture and the supernatant was collected in a fresh tube.
·
100 µl of 5M sodium acetate was added to
the contents and was mixed gently.
·
2 ml of isopropanol was added and mixed
gently by inversion till white strands of DNA precipitates out.
·
The contents were centrifuged at 5,000
rpm for 10 minutes.
·
The supernatant was removed and 1ml 70%
ethanol was added.
·
The above contents were centrifuged at
5,000 rpm for 10 minutes.
·
After air drying for 5 minutes 200 µl of
TE buffer or distilled water was added
·
10 µl of DNA sample was taken and was
diluted to 1 or 2 ml with distilled water.
·
The concentration of DNA was determined
using a spectrophotometer at 260/280 nm.
·
The remaining samples were stored for
further experiments.
Precautions:
·
Cut tips should be used so that the DNA
is not subjected to mechanical disruption.
·
Depending on the source of DNA the
incubation period of Proteinase K should extended.
·
The phenol chloroform extraction should
be repeated depending on the source of DNA to obtain pure DNA.
·
DNase free plastic wares and reagents
should be used.
Result
The sample should be observed in a UV visible spectrometer for its
absorbance maxima. If the sample obtains a peak at the range of 260 nm then it
is confirmed that the sample contains DNA in it.
Comments
Post a Comment